

Update Post: A Timeline of Hemophilia Research
Posted by Wendy Wise on
Hemophilia is a bleeding disorder in which the body is unable to form blood clots properly, which can lead to problems like spontaneous bleeding and excessive bleeding upon injury. This disorder is caused by a genetic deficiency in one of the proteins associated with the coagulation cascade, a series of reactions that leads to the formation of a stable blood clot. When the body is attempting to form a clot, this cascade comes to a halt when the protein responsible for the next step in the chain is missing; as a result, the clot is unable to form as it normally would.
Types of Hemophilia and Inheritance Patterns
There are several known forms of hemophilia, some more common than others, and each associated with the lack of a different coagulation-related protein.
- Hemophilia A is associated with a lack of Factor VIII (most common form of hemophilia)
- Hemophilia B is associated with a lack of Factor IX (less common form of hemophilia)
- Hemophilia C is associated with a lack of Factor X (least common, least severe form of hemophilia)
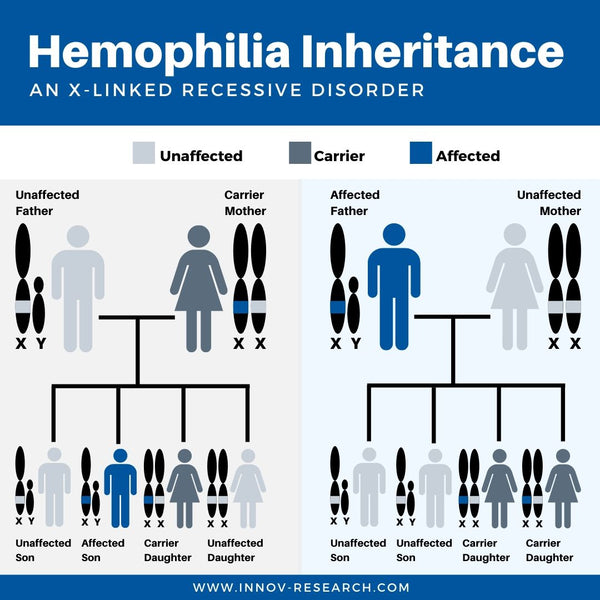
Hemophilia A and B are both X-linked recessive disorders, which means that the gene for these factors is carried on the X chromosome. A male, having one X and one Y chromosome, needs to inherit a disordered gene from his mother to have the condition. A female, having two X chromosomes, would need to inherit two copies of the hemophilia gene (one from each parent). Because hemophilia A and B are X-linked recessive disorders, these conditions almost exclusively affect male offspring.
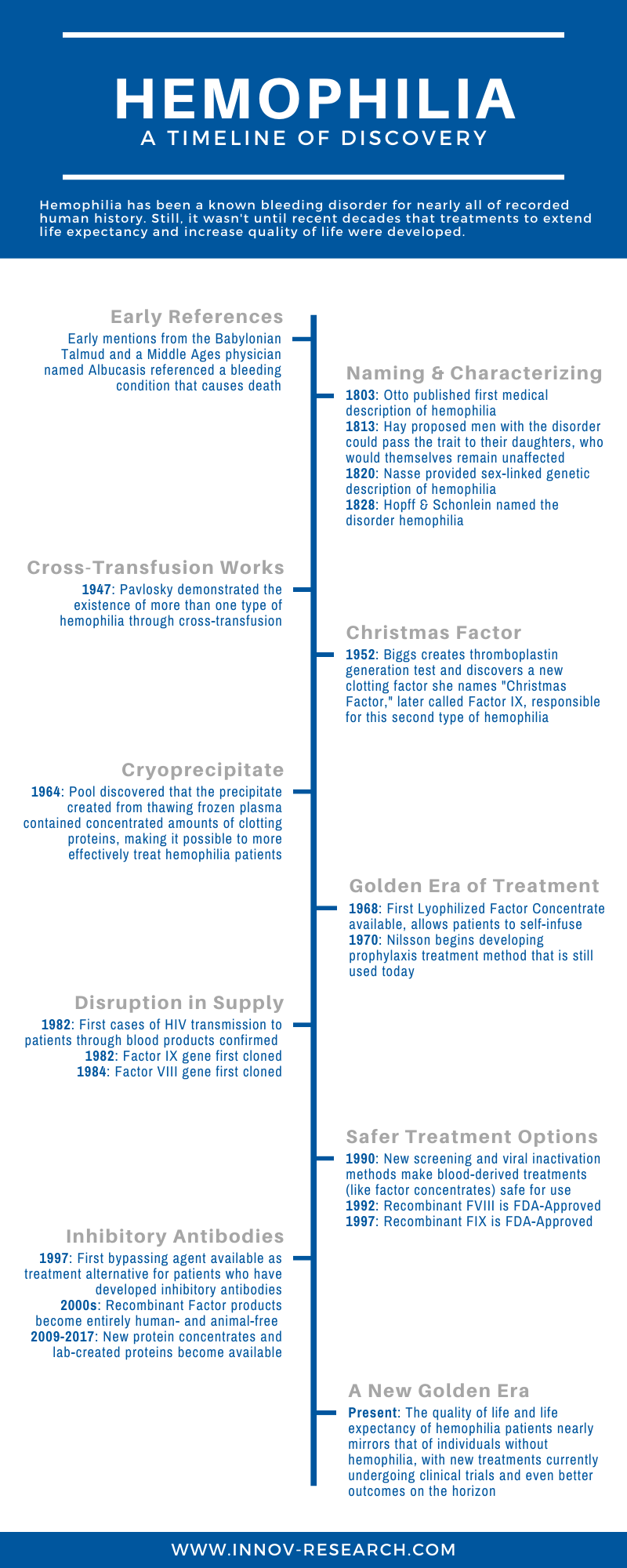
Until relatively modern times not much was known about the cause of this disorder, even though the symptoms have been described in written history for millennia. How did we go from simply knowing the symptoms exist to fully characterizing and treating the disorder? It’s a long and fascinating journey, with significant progress made in modern history that is creating more promising patient outcomes all the time.
Recognizing and characterizing the condition
Early references to hemophilia have been found dating as far back as the 2nd century AD. The Babylonian Talmud asserts that a male infant should not be circumcised if two of his brothers have previously died as a result of excessive bleeding from the procedure. In the 12th century, physician Abulcasis, known as the greatest surgeon of the Middle Ages and often credited as the “father of surgery,” described a family in which males died from bleeding upon trivial injury. In the early years of the 19th century, many academic papers were written in which physicians and scientists were able to determine the sex-linked pattern of genetic inheritance for hemophilia, and the bleeding disorder was officially named.
Developing a deeper understanding
While the pathophysiology of the disorder was becoming increasingly understood, a bigger question began developing as to whether this was a single disorder or if there could be more than one cause of the condition. In 1947, Dr. Alfredo Pavlovsky demonstrated that a blood transfusion from one hemophilia patent was able to correct the clotting problem in a second patient, and that the reverse was also true (a transfusion from the second to the first also worked). These results suggested that there may, in fact, be two distinct forms of hemophilia.
Meanwhile, Dr. Rosemary Biggs and her team at Oxford had designed the thromboplastin generation test in 1952, which could detect specific blood defects among hemophilia patients. In the course of their research, they discovered that a patient named Stephen Christmas did not demonstrate the characteristic Factor VIII deficiency associated with hemophilia but instead lacked a previously unknown clotting enzyme that they named Christmas Factor. With this finding, there existed two distinctly recognizable forms of hemophilia. Hemophilia A was proposed to refer to the more common Factor VIII deficiency, while hemophilia B was proposed for this less common Christmas Disease, later identified to be a specific deficiency in Factor IX.
While hemophilia was recognized, named, and demonstrating a known pattern of inheritance by the turn of the 20th century, there were still no real treatments for the disorder aside from direct blood or plasma transfusions. While this was the most effective known treatment through the 1960s, it was of limited effectiveness as the overall concentration of Factor VIII or Factor IX in blood and plasma is too low to stop severe cases of bleeding. Because of this, most hemophilia patients died in childhood or early adulthood from bleeding related to trauma or from hemorrhages in vital organs, especially the brain. As late as 1960, the life expectancy for a patient with severe hemophilia was less than 20 years of age.
The ”Modern” Age of Hemophilia Treatment Begins
Dr. Judith Graham Pool, a scientist and researcher, made a groundbreaking discovery in 1964 that changed the course of hemophilia treatment. She found that the precipitate left from thawing plasma (cryoprecipitate) contained significant amounts of Factor VIII. This was an enormous step forward in hemophilia treatment, as it was now possible for blood banks to create and store this component directly. This cryoprecipitate could be given to hemophilia patients in relatively small volumes to help with clotting during bleeding episodes. It also was the start of prophylaxis (preventative treatment) for hemophilia, as the cryoprecipitate could be given in advance of surgery, making emergency and elective surgery a possibility for the first time.
By the 1970s, the era known as “modern” treatment of hemophilia was in full swing. Lyophilized concentrates of Factor VIII and Factor IX were widely available, revolutionizing hemophilia care as patients were able to store concentrates in their own homes and “self-infuse” factor products when needed. This had the benefit of reducing the need for a hospital trip for treatment, and because these factor concentrates could be administered immediately, patients could take early control of bleeding episodes. Not only that, but elective surgery was also safe for hemophilia patients, including corrective surgery to alleviate issues caused by prior hemorrhage. This greatly improved both quality of life and life expectancy for hemophilia patients.
In the meantime, Dr. Inga Marie Nilsson was hard at work in Sweden. By the 1970s, she was developing a primary prophylaxis treatment in which hemophilia patients received factor infusions on an ongoing, preventative basis instead of postponing treatment until hemorrhagic incident. She was able to demonstrate that this strategy, along with active orthopedic measures, could prevent hemorrhage and ward off other problems like joint destruction, muscle atrophy, and disability. While her work was controversial in the beginning, this model is now widely accepted as the best course of action for hemophilia patients during periods of active growth.
Treatment Safety in the Age of HIV & Hepatitis C
This so-called “golden era” of hemophilia treatment came to a rapid halt in the 1980s when HIV and Hepatitis C were confirmed to be transmitted by blood and blood products, which had a devastating impact on the hemophilia community. Half of all hemophilia patients in the US contracted the HIV virus, and 44% contracted Hepatitis C. Traditional factor concentrates became safer in the 1990s, as screening became stronger and advanced methods of viral inactivation were used to produce safe plasma-derived factor products. There have been no reports of blood-borne transmission of hepatitis or HIV viruses in more than 30 years.
Other scientific advances that began in the 1980s were becoming the innovative treatments of the 1990s. In the 1980s, the Factor VIII and Factor IX genes were cloned, which paved the way for industrial production of recombinant factor proteins. The first recombinant Factor VIII product was approved by the FDA for therapeutic use in 1992, and the recombinant Factor IX was FDA approved in 1997.
A New Side Effect: Inhibitory Antibodies
With these advances in treatment came a new occurrence; some hemophilia patients developed inhibitory antibodies to infused factor product which prevented the treatments from working. In the 2000s, research in this area advanced rapidly and recombinant factor products became entirely free of human and animal derivatives, which lowered the risk for inhibitor development. The introduction of immune tolerance induction, a long-term IV infusion of large doses of coagulation factors, has allowed for the eradication of inhibitors in nearly two-thirds of patients while decreasing frequency of treatment from 2-3 times per week to once per week or less. Today, all licensed coagulation factors – both plasma-derived and recombinant – are highly effective with high safety and success rates.
A New “Golden Era” of Hemophilia Treatment
In the past decade, high-quality factor replacement therapy has become widely available and has ushered in a new golden era in hemophilia treatment. The quality of life and life expectancy of patients has greatly increased, and in high-income countries now nearly matches that of males without hemophilia. The most challenging problem of hemophilia treatment today is the development of inhibitory alloantibodies against Factor VIII or Factor IX, affecting 25%-30% of patients with severe hemophilia A and 3% to 5% of patients with hemophilia B. There are several promising pharmaceutical treatments to solve this currently undergoing clinical trials.
The availability of modern technology has rapidly and dramatically changed the course of hemophilia treatment in the past few decades alone. While there is no absolute cure yet, pharmacological research and new clinical studies on gene therapy are incredibly promising, and the coming decade is sure to see its own share of incredible advancement.
Further Reading & References:
- Franchini, M., & Mannucci, P. (2014). The History of Hemophilia. Seminars in Thrombosis and Hemostasis, 40(05), 571–576. doi:10.1055/s-0034-1381232
- Holmberg, L. (2000). In Memoriam, Inga Marie Nilsson, 1923–1999. Thrombosis Research, 98(4), 229–230. doi:10.1016/s0049-3848(00)90000-8
- History of Bleeding Disorders. National Hemophilia Foundation. https://www.hemophilia.org/Bleeding-Disorders/History-of-Bleeding-Disorders
- NIH U.S. National Library of Medicine, Genetics Home Reference. https://ghr.nlm.nih.gov/condition/hemophilia
Interested in Hemophilia Research?
Innovative Research offers a wide range of coagulation-related products to support many different areas of research.